How to Get FDA Approval for Your Startup in 6 Steps
Without approval from the Food and Drug Administration (FDA), you can’t market your medical product in the U.S. Getting FDA approval is a challenging obstacle on the path to market for a medical startup, but it’s well worth the effort.
FORGE invited Iris Sherman, biosecurity and population research group lead at MITRE, to share her wisdom with the attendees at our webinar Demystifying Medical Device Classification & Approval with MITRE. Sherman began her career in the medical innovation space. Ever heard of Allegra? Sherman was on the drug’s founding team. She later worked with the FDA to build a medical device approval process for sub-Saharan Africa. Sherman knows the ins and outs of the regulatory process for medical devices.
Here’s what you need to know about FDA approval.
Know your device’s class
Plenty of companies go bankrupt while following the FDA’s premarket notification process, also known as a 510(k), when they should have gone through the De Novo or Pre-Market Approval process instead. For the best chance of success, first determine what FDA class your device falls into, and which submission pathway to follow.
The FDA groups medical products in three classes based on risk to the patient. A Class I device could be something simple and external, like a bandage. Class II have a moderate to high risk, like a power wheelchair. A pacemaker supports a major body function, so it would be in Class III. The FDA has resources to help you decide what class your product belongs to. It’s simple enough that you can do it yourself.
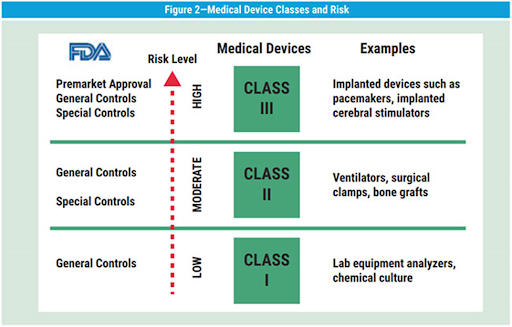
Choose the right type of submission
- Even for the least risky class, medical device companies need to show the FDA that their devices are safe. Once you know which of the three classes is the right one for your device, you can prepare the appropriate regulatory submission. Applying for a 510(k) approval requires proof that your device is substantially equivalent to an existing product, called a predicate device. Most people consider this the easiest path to market. However, it takes a lot of work to prove your device is comparable to the predicate.
- The most stringent process, Pre-Market Approval (PMA), is for Class III devices. Companies must have valid scientific evidence of safety, including clinical laboratory and human testing.
- If you have a device unlike anything else on the market, a De Novo submission lets you start with a clean slate. It’s like a 510(k), but you don’t have to prove your device is similar to a predicate device.
Know your market
It’s good to double-check, but most devices fit the 510(k) pathway. That means finding the right predicate device as a comparison point. Use your competitors as examples. Their devices made it through FDA approval because their applications met the FDA’s strict guidelines.
Look up previous 510(k) submissions on the FDA’s database. You’ll find data like the manufacturer’s name and the key number. Google the key number and you’ll often find a device’s full 510(k) executive summary. This document goes beyond the intended uses and substantial equivalence to give you the exact tests that company ran. Don’t be afraid to copy language from your predicate device’s documentation verbatim. It’s still up to you to prove the claims about your device, though.
Flow chart: Is the predicate device you’ve chosen “substantially equivalent” to your product?
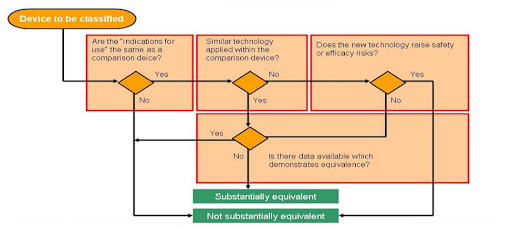
Document thoroughly for FDA approval
The FDA wants to prevent recalls, so they ask for a lot of documentation. Starting early in the design and development process, build a dossier on your device. Keep notes on every element of your prototype, along with blueprints, test results, and changes you make during development.
You must list every possible malfunction or user error during the regulatory process. The FDA needs to know you can address any future issues and trace their cause.
Collaborate with the FDA
It’s best practice to apply for an “Q-submissions meeting” (Q-sub) with FDA reviewers.During Q-sub meetings, you can provide an overview of your device development and familiarize reviewers with the significance of your device. The meeting is a valuable opportunity to discuss your submission and get advice about how to win FDA approval. Proactive FDA interaction will save you time down the road.
Here are some good questions to ask at a Q-sub meeting:
- Are the proposed trial design and selected control group appropriate?
- Does the FDA concur with our alternative test method? It’s not the recognized standard.
- Is a “moderate level of concern” the appropriate level of concern for my software?
- Are there concerns with the predicate device proposed?
- What specific information about a post-approval study should the PMA contain?
- Are the proposed study designs for demonstrating precision and accuracy adequate to support use of the test in the Phase 3 clinical study?
The FDA won’t give you feedback during the meeting, but you can submit a written formal request for feedback.
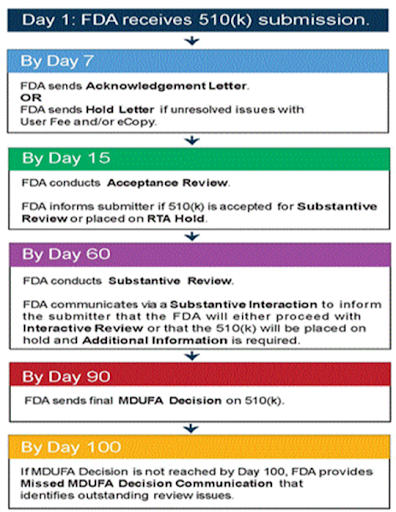
Prepare for a long process
FDA approval is a lengthy process. If every step of the 510(k) submission process goes perfectly and there are no questions, it will take 100 days. It often takes longer. Sherman suggests preparing for a timeline of 150 days. Be sure to inform collaborators of the expected timeline before submitting.
Mistakes will affect your FDA review, so it helps to get it right the first time. Writing for the FDA requires a unique skillset, so if you feel uncertain about your submission, it’s not a bad idea to engage an FDA submission expert to compile and/or review it. Need assistance locating one? FORGE can help!
And don’t forget – it’s against regulations to promote your device before it’s approved! Reviewers often visit company websites to learn about a company and its device. Be patient and hold off on the marketing until the FDA gives you approval.
Links you need to submit your medical device to the FDA
These FDA resources will help you classify your device, find predicate devices, and prepare a submission.
- How to classify your medical device
- Regulatory controls
- Class I/II exemptions
- Premarket 510(k) overview
- Premarket approval overview
- 510(k) clearance database
- Premarket Approval (PMA) database
- Establishment Registration and Device Listing
- De Novo database
- Medical device classification product codes – guidance for industry and FDA staff
- FDA and industry procedures for section 513(g) requests for information under the Federal Food, Drug, and Cosmetic Act
- Cybersecurity Toolkit for Digital Health
We hope Sherman’s expertise helps guide you through the FDA approval process. Submitting your device for approval can be time-consuming, stressful, and expensive, but it’s required to sell your device in the U.S. and is an exciting step on the journey to commercialization. You can do it!
Here are the slides and video recording from the event. Sign up for FORGE’S newsletter to be informed about future event opportunities for medical devices and beyond.